


BLAST(Basic Local Alignment Search Tool)是生物信息学中最为重要的序列相似性搜索工具之一,广泛应用于DNA、RNA及蛋白质序列的相似性比较,BLASTn和BLASTp作为BLAST工具中的两个主要程序,各自扮演着不同但互补的角色,本文旨在详细解析BLASTn与BLASTp的区别,并探讨它们在生物信息学研究中的应用。

基本原理的差异
1、BLASTn的基本原理
BLASTn专门用于核酸序列间的比对,它接受DNA或RNA序列作为查询条目,并将其与核酸数据库中的其他序列进行比较,以找到相似的序列,这个过程涉及检测查询序列与数据库序列之间的匹配程度,并评估这些匹配的统计显著性。
由于BLASTn直接作用于核酸序列,它不会涉及任何翻译过程,这使得它非常适合于基因组注释、物种鉴定和寻找同源基因等需要直接核酸比对的场景。
2、BLASTp的基本原理
相反,BLASTp被设计用于蛋白质序列之间的比对,它将查询的蛋白质序列与蛋白质数据库中的序列对比,寻找相似的蛋白质,这个比对过程考虑了氨基酸的替代矩阵,反映了蛋白质层面进化过程中的变化。
BLASTp适用于寻找同源蛋白质、预测蛋白质功能和结构以及识别蛋白质家族等方面的研究,通过这种方式,研究人员可以探索不同物种中相同或相似功能蛋白质的分布及其演化关系。

输入类型与应用范围
1、BLASTn的输入类型与应用场景
BLASTn仅接受核酸序列作为输入,将其与核酸数据库中的序列进行比较,这种比对对于研究基因组结构、注释新发现的基因、识别基因中的SNPs(单核苷酸多态性)及Indels(插入/缺失)等具有重要意义。
2、BLASTp的输入类型与应用场景
BLASTp专门处理蛋白质序列,用以搜索和比对蛋白质数据库,它广泛应用于蛋白质的功能和结构预测、同源蛋白的搜索以及蛋白质进化分析等领域,尤其是在蛋白质工程和系统生物学研究中,BLASTp是不可或缺的工具。
数据解读与结果解释
1、BLASTn的结果解释

BLASTn的结果通常提供了匹配序列的相似性百分比、E值(期望值)、得分等统计参数,帮助研究者评估匹配的显著性,高相似性和低E值通常指示着强烈的同源性信号,这对于基因组学研究尤其重要。
2、BLASTp的结果解释
BLASTp的结果侧重于展示蛋白质序列之间的相似性和同源关系,它提供的输出包括匹配区域的氨基酸替换、插入或缺失等信息,这对于了解蛋白质的结构和功能变异至关重要。
综合以上各方面,可以看到BLASTn和BLASTp在生物信息学领域各自有着明确且独特的作用和应用,尽管两者都旨在通过序列比对揭示生物学信息,但它们的侧重点和应用背景大不相同。
在选择使用BLASTn还是BLASTp时,应基于具体的研究目标和可用数据类型做出决定,这两种工具为生物学家提供强大的支持,帮助他们解析遗传信息的复杂性,推动生命科学的发展。
相关问答FAQs
Q1: 是否可以将核酸序列直接用BLASTp进行比对?
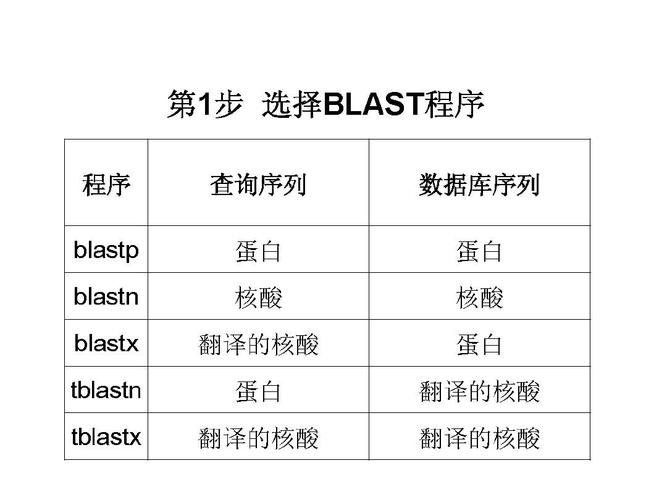
Q2: 如何选择合适的BLAST程序进行序列比对?
A2: 选择BLAST程序主要取决于待分析序列的类型(核酸或蛋白质)以及研究目的,如果是进行基因发现、基因组注释或种属鉴定,通常选用BLASTn;若目的是研究蛋白质功能、家族归类或结构预测,则应选BLASTp,若需从未知DNA序列中寻找编码蛋白质的区域,可考虑使用BLASTx或tblastx,明确研究需求和序列类型是选择BLAST程序的关键。
BLASTn和BLASTp虽然都是BLAST工具的一部分,但它们在原理、应用和结果解释上有着本质的不同,理解这些差异对于进行生物信息学研究至关重要,希望本文能为研究人员在选择和使用这些工具时提供指导。
原创文章,作者:未希,如若转载,请注明出处:https://www.kdun.com/ask/919069.html
本网站发布或转载的文章及图片均来自网络,其原创性以及文中表达的观点和判断不代表本网站。如有问题,请联系客服处理。
发表回复